



Kawasaki disease (KD), also known as mucocutaneous lymph node syndrome, is an acute, self-limited vasculitis that predominantly affects children, typically under the age of 5 years. It is the leading cause of acquired heart disease in children in developed countries, and its diagnosis and management are of paramount importance in pediatric practice. Kawasaki disease can lead to significant cardiovascular complications, particularly coronary artery aneurysms, which makes early identification and treatment critical.
This article provides a comprehensive review of the epidemiology, pathogenesis, pathophysiology, differential diagnoses, clinical features, diagnosis, treatment, and potential complications of Kawasaki disease.
Epidemiology
Kawasaki disease is most commonly seen in children under the age of 5, with the highest incidence occurring in toddlers. The incidence varies geographically, with higher rates reported in Japan and other East Asian countries. In Japan specifically, the incidence is approximately 200 cases per 100,000 children under the age of 5 years. In the United States, the incidence is estimated at around 19–25 cases per 100,000 children under the age of 5, with a slight male predominance (1.5–2:1 ratio). However, the disease can occur in children of any age, and there is a biphasic age distribution, with a second smaller peak in older children (ages 6–10 years).
Although the exact etiology of Kawasaki disease remains unclear, several risk factors have been identified:
- Genetic predisposition: Certain genetic markers have been linked to an increased risk of developing KD, with other markers being linked to higher rates of responsiveness to IVIG, and another subset of markers associated with higher rates of negative coronary artery outcomes.
- Ethnicity: Kawasaki disease has a strong association with Asian ancestry, particularly in Japanese and Korean populations. However, it has been reported in all ethnic groups worldwide. Risk is also more associated with ethnicity rather than geographic location, for example those in Hawaii have a similar risk to those in China rather than the rest of the United States.
- Infectious triggers: While no specific infectious agent has been consistently identified, there is some evidence suggesting that seasonal variations and geographic clustering in viral infections may indicate an infectious trigger in certain subsets of the population more genetically at risk.
Pathogenesis and pathophysiology
The pathogenesis of Kawasaki disease is believed to involve an immune-mediated response triggered by an environmental or infectious factor in genetically predisposed individuals. Although the exact trigger remains unknown, the immune system appears to react inappropriately, leading to widespread inflammation of small and medium-sized blood vessels.
The acute phase (first two weeks) is mediated by an innate immune response where neutrophilic processes cause lysis of cells at the arterial wall level, leading to aneurysm formation and loss of vascular wall integrity.
The subacute phase follows in the weeks to months after, consisting of a mainly acquired immune response heave on lymphocytes, plasma cell, and eosinophilic mediated damage.
More long-term sequelae, over months to years, is categorized in a luminal myofibroblastic proliferation phase where there is vascular stenosis and changes to the changes in blood flow pattern from laminar to more circular flow. This phase is thought to contribute to the remote complications of cardiomyopathy, arrhythmias, and heart failure for these patients in early adulthood, including in those patients without a history of aneurysm formation in their acute/subacute phases.
The hallmark of Kawasaki disease is its ability to cause systemic vasculitis, which affects various tissues, particularly the coronary arteries. The most severe complication of KD is the formation of coronary artery aneurysms (CAAs), which can lead to long-term cardiovascular morbidity, including myocardial infarction, arrhythmias, and sudden death.
Other vascular changes can include dilatation of the aorta and involvement of the peripheral arteries, although these are less common.
Differential diagnosis
Several conditions can mimic the clinical presentation of Kawasaki disease. A thorough differential diagnosis is important to avoid misdiagnosis and to ensure appropriate management.
- Scarlet fever: Caused by Group A Streptococcus, it can present with a rash, lymphadenopathy, and fever similar to KD. However, they often lack the longer duration of fever and if conjunctival injection is present it tends to be exudative.
- Viral infections: Conditions such as measles, adenovirus, and enteroviral infections can present with prolonged fever and a rash.
- Toxic shock syndrome: Caused by Staphylococcus aureus or Streptococcus pyogenes, this condition presents with fever, hypotension, refractory tachycardia, and a rash. Although it may have some other ancillary symptoms, and can mimic Kawasaki shock syndrome, these patients often don’t have the other clinical criteria specific to KD.
- Rheumatologic:
- Systemic juvenile idiopathic arthritis.
- Systemic lupus erythematosus.
- Drug reactions:
- Stevens-Johnson Syndrome.
- Erythema Multiforme Major.
- Serum sickness/Serum sickness-like reaction (higher suspicion with recent drug use).
Clinical features
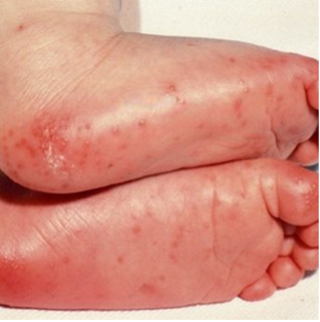
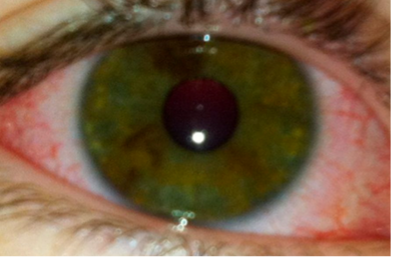
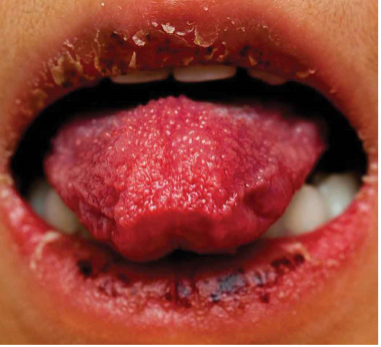
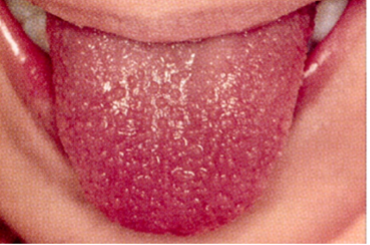
CRASH and BURN: Conjunctivitis, Rash, Adenopathy, Strawberry tongue, Hands and feet, and BURN (fever ≥ 5 days) are the most common features of Kawasaki disease.
Diagnosis
Kawasaki disease is diagnosed clinically based on the presence of fever for at least 4 days and at least four of the following five clinical features:
- Bilateral conjunctival injection (non-exudative, often painless but itchy). Often limbic sparing as the area surrounding the iris is an avascular zone. Uveitis can be used to meet this criteria is seen on slit lamp.
- Oral mucosal changes: Redness and fissuring of the lips, strawberry tongue (which references hypertrophy of the papillae and does not require associated erythema to diagnose), and oral mucosal erythema. Ulcerations and exudates are not common.
- Polymorphous rash: Most fluctuating symptom. No specific phenotype; however, is not bullous, petechial, or vesicular.
- Cervical lymphadenopathy (usually >1.5cm in diameter).
- Peripheral extremity changes: Erythema, swelling, or desquamation.
- If four out of five of the clinical criteria are not met, classic Kawasaki disease cannot be diagnosed. However, there is still the opportunity to diagnose incomplete Kawasaki disease when there are at least two clinical criteria present with either an abnormal ECHO or at least three supplemental lab criteria.
- The clinical symptoms do not need to be present simultaneously, making a good history of the past few weeks incredibly important.
- Elevated inflammatory markers: Increased C-reactive protein (CRP) and/or erythrocyte sedimentation rate (ESR).
- Leukocytosis with WBC >15.
- Anemia for age.
- Albumin ≤3.
- Platelets >450,000 after day 7 of illness (this is a delayed phase reactant and thus lower levels early in illness should not be interpreted with a grain of salt).
- Elevated ALT for age.
- At least 10 WBC/hpf from urinalysis (this should be a bag specimen as the WBC in questions are from urethritis and a catheter specimen may bypass the affected area and yield falsely normal results).
- Echocardiography: This is performed on initial diagnosis to both evaluate for small vessel abnormalities, but also to get a baseline for cardiology to compare with over time as the higher risk for cardiac abnormalities often comes later in the course.
- There will be repeat ECHOs done at 1–2 weeks and ~6 weeks from initial presentation in all patients, with higher frequency for those with abnormalities already present.
- The one measurement to take note of on the ECHO report will be the z-score: Normal is <2, dilatation is 2–2.5, small aneurysms are 2.5–5, medium aneurysms are 5–8, and large are >8.
- CTA and Cardiac MRI can also be used for better resolution images and better views of possible peripheral artery stenosis; however, they are higher cost and have the risks associated with anesthesia.
Treatment
The mainstay of treatment is IVIG, which is given as a single dose of 2 g/kg over 12-24 hours (it can cause hypotension when run too quickly). IVIG has been shown to reduce the incidence of coronary artery abnormalities and is most effective if given within the first 10 days of illness. However, if given too early (within the first five days of illness) there is a slight increase to the rates of “refractory KD” and patients requiring a second IVIG.
Notes on IVIG:
- It is a pooled blood product, so is contraindicated in IgA deficiency due to the risk of overwhelming anaphylaxis.
- Lives vaccines need to be deferred for at least 11 months post-IVIG due to concern about a lack of robust immune response. They can be given earlier if there is a specific outbreak or patients are living/traveling to an endemic area, however they will need another dose after that 11 months to count as part of the recommended regimen.
- IVIG causes increases to the ESR, thus ESR should not be used for diagnostic purposes for at least 1 month post-infusion.
Aspirin is used in two phases of treatment:
- High-dose aspirin (80–100 mg/kg/day in the United States, but 30-50mg/kg/day in Asian countries) during the acute phase (first 24–48 hours) for its anti-inflammatory effects.
- Low-dose aspirin (3–5 mg/kg/day) is used for the next 6 weeks for its antiplatelet effect.
Notes on Aspirin:
- NSAIDs should be avoided while in aspirin as they can interfere with the antiplatelet effects.
- Patients with G6PD deficiency should only get low dose aspirin to lower the risk of a hemolytic anemia flare.
- Reye’s syndrome is liver injury that can occur in children in the setting of aspirin use. Clinical findings include tachypnea, abdominal pain, vomiting, diarrhea, altered mental status, seizures, paralysis, liver and kidney failure. Lab findings include elevated LFTs, prolonged prothrombin time, metabolic acidosis, respiratory alkalosis, elevated ammonia, and hypoglycemia.
- The risk of Reyes is highest in the setting of flu and varicella, thus in patients with KD who have had either of those infections within the past 6 weeks the care team may choose to use steroids in places of aspirin for the anti-inflammatory effect.
Steroids are not routinely used but may be considered in patients with resistant Kawasaki disease (those who do not respond to initial IVIG treatment), those with contraindications to aspirin therapy, or in patients with significant cardiovascular involvement.
In refractory cases, additional treatments, including certain monoclonal antibodies may be used.
Potential complications
Kawasaki disease, if untreated or inadequately treated, can lead to significant complications:
- Coronary artery aneurysms: The most serious complication, which may lead to myocardial infarction, arrhythmias, or sudden death.
- Myocarditis (more common): Inflammation of the heart muscle can occur and contribute to heart failure in the acute setting.
- Pericarditis (less common): Inflammation of the pericardium can cause chest pain and effusion.
- Thrombosis: The formation of blood clots in the coronary arteries can be a later consequence of aneurysm formation.
- Long-term cardiovascular sequelae: Children who survive KD may develop long-term coronary artery disease, requiring lifelong monitoring.
Conclusion
Kawasaki disease is an acute vasculitis that can lead to severe complications, particularly involving the coronary arteries. Early diagnosis and treatment, including intravenous immunoglobulin and aspirin, are critical to reducing the risk of coronary artery aneurysms and other cardiovascular complications. Pediatricians and healthcare providers should remain vigilant for the signs and symptoms of KD, particularly in children presenting with prolonged fever (4 or more days) and characteristic clinical features. Early intervention significantly improves outcomes and reduces long-term cardiovascular morbidity.
- McCrindle, B. W., et al. (2017). Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: A Scientific Statement from the American Heart Association. Circulation, 135(17), e927–e999.
- Son, M. B., & Tremoulet, A. H. (2016). Kawasaki Disease: Insights into Pathogenesis and Treatment. The Journal of Pediatrics, 175, 1–8.
- Newburger, J. W., et al. (2017). Kawasaki Disease: Clinical Features, Diagnosis, and Management. Pediatric Clinics of North America, 64(4), 651–671.
- Kawasaki, T., et al. (1967). A New Acute Febrile Mucocutaneous Lymph Node Syndrome with Exanthema in Children. The Journal of Pediatrics, 71(1), 1–17.
- Ronai, Christina & Hamaoka-Okamoto, Akiko & Baker, Annette & Ferranti, Sarah & Colan, Steven & Newburger, Jane & Friedman, Kevin. Coronary Artery Aneurysm Measurement and Z Score Variability in Kawasaki Disease. Journal of the American Society of Echocardiography, Volume 29, Issue 2,
2016, Pages 150–157, ISSN 0894-7317. - Jone, Pei-Ni., et al. (2024). Update on Diagnosis and Management of Kawasaki Disease: A Scientific Statement from the American Heart Association. Circulation. 150, 23.