- Suggested reading
- Reading guide
- Need to Know
There is one chapter for suggested reading from Lippincott, Chapter 20: Amino Acids: Degradation and Synthesis. The short descriptions listed below are some of the big-picture, high-yield points. The reading and the class session will go into more depth and detail. The reading guides are also posted.
As I have said in other modules, this reading guide is for people who need help focusing on these chapters. Most students DO NOT need to use the guide.
Read Chapter 20.
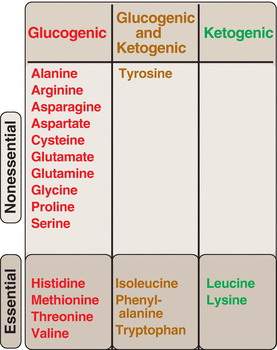
The carbon skeleton of amino acids is broken down to glucogenic and ketogenic compounds
-
Big picture
-
What does the body do with the breakdown products of the carbon backbone of amino acids?
-
What’s the definition of a glucogenic amino acid?
-
What is the definition of a ketogenic amino acid?
-
Can an amino acid be both glucogenic and ketogenic?
-
What is the definition of an essential amino acid?
-
-
If you understand and reproduce the figure on the previous page, you can answer all the following questions:
-
Which amino acids are glucogenic? (Fig. 20.2)
-
Which amino acids are ketogenic?
-
Which of the 20 amino acids incorporated into protein are considered essential amino acids?
-
Which amino acids degrade to oxaloacetate?
-
Which amino acids degrade to a-ketoglutarate?
-
Which amino acids degrade to pyruvate?
-
Which amino acids degrade to fumarate?
-
Here’s a tricky question: Which essential amino acid, in addition to threonine, ultimately degrades to pyruvate?
-
Which amino acids break down to acetoacetate?
-
Which amino acids break down to acetyl-CoA?
-
Which amino acids breakdown to both acetyl-CoA and acetoacetate?
-
-
Breakdown (and resynthesis) of methionine
-
The metabolism of methionine is important because of its role in the generation of S-adenosylmethionine (SAM), a potent methyl donor molecule, and the production of homocysteine.
-
How is methionine converted to homocysteine?
-
Into what two primary products can homocysteine converted?
-
Which amino acid is converted to cysteine when synthesized from cystathionine? Homocysteine or serine?
-
What two conditions, named in Harvey’s, are associated with elevated circulating homocysteine levels?
-
What kind of deficiencies could lead to elevated homocysteine?
-
-
The branched chain (essential) amino acids
-
These amino acids are deaminated by the same branched chain-aminotransferase and then decarboxylated by the same branched-chain a-keto acid dehydrogenase complex.
-
Which of the branched chain amino acids is only glucogenic?
-
Which of the branched chain amino acids is only ketogenic?
-
What enzyme deficiency leads to maple syrup urine disease?
-
Why does the urine smell like maple syrup?
-
The synthetic importance of folic acid (Fig. 20.11)
-
Why is folate (specifically tetrahydrofolic acid; THF) important (big picture question)?
-
How does folate deficiency often present?
The synthesis of nonessential amino acids
-
These are the 11 amino acids mainly from the upper part of the figure on page 2. Their synthesis is straight forward, compared to their degradation. The figure on the following page contains most of the details you’ll need to know. Notice the central role played by glutamate in the synthesis of several amino acids. Some of this you’ve already seen before (i.e. amidation of asp and glu).
-
Which amino acids are ultimately made from a-ketoglutarate?
-
What other amino acids are directly or indirectly synthesized from intermediates of metabolism?
-
Which amino acids are made from essential amino acids?
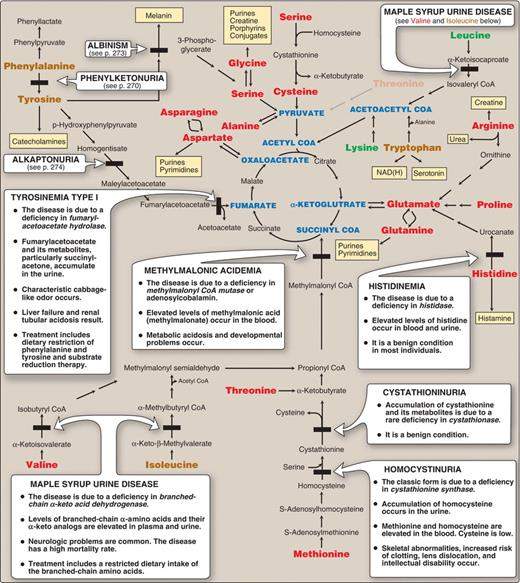
Metabolic diseases caused by metabolic defects in amino acid metabolism
-
They are rare but there are a lot of them. They can be devastating and usually need to be diagnosed early to optimize the outcome for the patient.
-
Phenylketonuria (PKU) (Fig. 20.15,17)
-
What causes PKU?
-
What other reactions involving amino acids are inhibited with BH4 deficiency? (Fig. 20.16)
-
What happens to patients with untreated PKU?
-
Why are PKU patients usually light colored?
-
When is the newborn screening for PKU done and why?
-
Can PKU be diagnosed prenatally? How about pre-implantation?
-
How is PKU treated?
-
When does treatment for PKU need to begin to avoid irreversible damage? (Fig. 20.18)
-
How long does treatment need to be continued? (Fig. 20.19)
-
Can maternal PKU affect a normal fetus?
-
-
Albinism (Fig. 20.20)
-
What is the general cause of albinism?
-
Besides lack of pigmentation in the hair, skin, and eyes, what other symptoms are routine in albinism?
-
-
Alkaptonuria (Fig. 20.22)
-
What causes alkaptonuria?
-
What are the symptoms of alkaptonuria?
-
What is the onset of symptoms in alkaptonuria?
-
-
Maple syrup urine disease (MSUD)
-
What causes MSUD?
-
What are the symptoms of MSUD?
-
What is the treatment for MSUD?
-
-
Homocystinuria (Fig. 20.21)
-
What causes homocystinuria?
-
What are the symptoms of homocystinuria?
-
What is the treatment for homocystinurea?
-
Summary
-
Metabolic diseases associated with amino acid metabolism (Fig. 20.14)
-
This is a very nice figure to review and get a sense of how all these metabolic diseases of amino acid metabolism relate to the larger metabolic picture. You do not, however, need to memorize this figure. Just remember the basics of the metabolic diseases related to amino acids that we have discussed here.
-
Describe the role of folic acid in amino acid metabolism
-
Tetrahydrafolate
-
“One carbon pool”
-
Methionine synthesis
-
Glycine biosynthesis
-
Serine biosynthesis
Diagram and describe how the different classes of amino acids are metabolized and what occurs (at the biochemical level) when there are defects in amino acid metabolism
-
Phenylalanine hydroxylase
-
α-keto acid dehydrogenase
-
Homogentisic acid oxidase
-
Alkaptonuria
-
Phenylketonuria
-
Homocystinuria
-
Maple syrup urine disease
-
Know what the product of the amino acids are (i.e. pyruvate, oxaloacetate, etc.) and how they relate to gluconeogenic or ketogenic substrates.
Describe the synthesis and catabolism of nonessential amino acids, including their precursors, enzymatic pathways, and metabolic fates
-
Essential amino acids
-
Nonessential amino acids
Describe the role of folic acid in amino acid metabolism
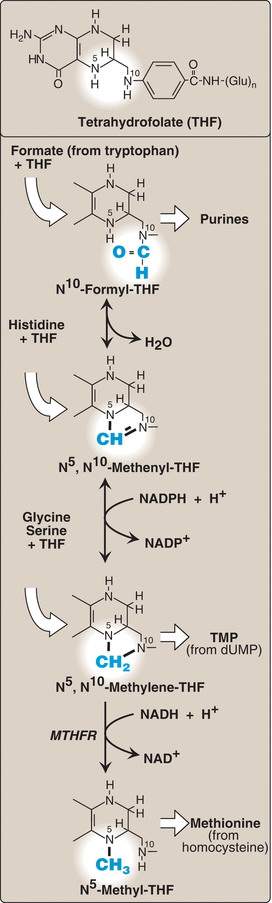
Folic acid is the synthetic form of folate, a water-soluble B vitamin that takes part in several critical functions in the human body. The active form of folate is tetrahydrofolic acid. It serves in one carbon reductions reactions. These reactions are involved in the synthesis of nucleotides and amino acids. The amino acids requiring folic acid for metabolism are methionine, cysteine, serine, glycine, and histidine. Folic acid also serves as a coenzyme in converting methionine to homocysteine.
Diagram and describe how the different classes of amino acids are metabolized and what occurs (at the biochemical level) when there are defects in amino acid metabolism
The big take-home here is that you need to know and understand phenylketonuria, maple syrup urine disease, and homocystinuria. See also this alkaptonuria video.
Describe the synthesis and catabolism of nonessential amino acids, including their precursors, enzymatic pathways, and metabolic fates
Note
Non-essential amino acids can be made by the body. Essential amino acids must come from the diet.
|
Synthesis from α-keto acids |
Alanine, aspartate, and glutamate |
Transamination reactions
|
|
Synthesis by amidation |
Glutamine and Asparagine
|
|
|
Related to on-carbon donors and one-carbon acceptors (think folic acid)
|
Serine, glycine, and cysteine
|
This relates to the first learning goal in this session.
|
|
From phenylalanine |
Tyrosine |
In patients with PKU, tyrosine becomes essential.
|
|
From glutamate |
Proline and arginine |
Note: Arginine can also be made via the urea cycle
|