


- Suggested reading
- Reading guide
- Need to Know
There is one chapter for suggested reading from Lippincott, Chapter 19: Amino Acids: Disposal of Nitrogen. The short descriptions listed below are some of the 20,000-foot picture, high-yield points. The reading and the class session will go into more depth and detail. The reading guides are also posted.
As I have said in other modules, this reading guide is for people who have difficulty focusing on these chapters. Most students DO NOT need to use the guide.
Read Chapter 19.
Proteins as the source and sink of amino acids
- Big picture (Fig. 19.2)
- How is the handling of amino acids by the body different from carbohydrates and lipids?
- Where does your body get amino acids and what is the amino acid pool?
- How big is the amino acid pool compared to the total protein in the body?
- What are the possible fates of amino acids (what are the options for exiting the amino acid pool)?
- Protein turnover
- On average, how much endogenous protein is broken down each day in normal, healthy individual?
- How much protein is synthesized in the same person?
- What control mechanisms (big picture) determines the levels of any given protein in the body?
- Converting endogenous proteins to amino acids (endogenous protein degradation) (Fig. 19.3 )
- What are the differences between the ubiquitin-proteasome proteolytic pathway and the lysosomal proteolytic pathway (energy requirements, source of proteins degraded by each)?
- What is ubiquitin and what happens to it as a result of proteasome action?
- Are all proteins equally likely to be degraded by the proteasome? What are some of the factors that determine a proteins half-life?
- Amino acids from dietary proteins (Fig. 19.4,5)
- What’s a zymogen?
- What protease is produced by the stomach and how is it activated?
- What are the primary proteases produced by the pancreas?
- What protease activates trypsinogen?
- What protease activates the other pancreatic proteolytic zymogens?
- What intestinal protease finishes the job that the pancreatic protease starts (that is, converts peptides into amino acids and dipeptides)?
- What protein breakdown products are ultimately transported to the liver from digestion?
- Amino acid transport
- How do cells take up amino acids from the extracellular space?
- What is the amino acid concentration difference between intra- and extra-cellular spaces, and how is it maintained?
- What is cystinurea and how does it relate to cellular amino acid transport mechanisms? (Fig. 19.6 )
Moving ammonia: The fate of amino acid nitrogen
- Big picture
- What is the major form of nitrogen released from amino acids?
- What is the major form of nitrogen secreted by the kidney?
- What two enzymes are required for the removal of the nitrogen from most amino acids? (Fig. 19.12)
- Transamination almost always involves one key amino acid and/or its respective a -keto acid. What amino acid and what a -keto acid?
- Transamination; moving amino groups to glutamate (Fig. 19.7)
- What two enzymes are required for the removal of the nitrogen from most amino acids?
- What is a “transamination” reaction?
- What amino acid are aminotransferases named after (amino donor or acceptor)?
- What molecule is typically the acceptor in the aminotransferase reaction?
(Fig. 19.7,8) - What coenzyme is required for transaminase reactions and from what vitamin is it derived? (Fig. 19.9)
- Are transamination reactions reversible?
- Which two aminotransferases are singled out as being particularly important and why? (Fig. 19.8,10)
- Oxidative deamination of amino acids
- What enzyme (and amino acid substrate) is most important in oxidative deamination?
- Are the reactions catalyzed by this enzyme reversible?
- What cofactor(s) does this enzyme require? (Fig. 19.11)
- What factors regulate this enzyme?
- Can we (humans) use the plant derived D-amino acids for protein synthesis?
- Transport of ammonia to the liver from other tissues (Fig. 19.13,18)
- What are the two mechanisms available for complexing ammonia for safe transport to the liver?
Urea cycle; safe disposal of ammonia
- Big picture
- What percentage of the nitrogen waste found in urine is in the form of urea?
- Where is the primary site of urea synthesis?
- What is the source(s) of the two nitrogen atoms in urea?
- How does urea get from the liver to the kidney for excretion?
- Why is glutamate central to the funneling of nitrogen to the urea cycle?
- The urea cycle (Fig. 19.14)
- From what amino acid is urea released?
- What amino acid is formed when urea is released from the amino acid you just named above?
- What are the four core intermediates in the urea cycle and how many are amino acids?
- What 4 enzymes are required to turn the urea cycle?
- What is the typical source of the ammonia for synthesis of carbamoyl phosphate?
- What enzyme catalyzes the formation of carbamoyl phosphate?
- What amino acid is added to citrulline to form argininosuccinate?
- From what intermediate is urea released?
- What steps of the urea cycle are energy dependent?
- What byproduct of the urea cycle can enter the TCA cycle?
- What reaction of the cycle is almost exclusive to the liver?
- What compounds regulate the urea cycle and how does this relate to what you eat? (Fig. 19.16)
- The fate of urea
- Where does most of the urea go?
- What happens to urea that finds its way to the intestines?
- What happens to urea in patients with kidney failure?
Urea good, ammonia bad
- Back to the big picture (Fig. 19.19)
- What are the major sources of ammonia (only a couple we haven’t talked about already)?
- What is the normal concentration range of ammonia in the blood?
- What happens when blood ammonia levels get too high?
- What are some common causes of acquired hyperammonemia?
- What is the most common genetic cause of hyperammonemia? (Fig. 19.20)
Summary (Fig. 19.21)
Describe the concept of the amino acid pool and compare and contrast amino acids acquisition from endogenous and exogenous protein sources
-
The amino acid pool
-
Zymogens
-
Trypsin
-
Elastase
-
Chymotrypsin
-
Intracellular protein degradation (Proteasome)
-
Dietary Protein Digestion
Describe ammonia transfer to and from amino acids and its transport and subsequent elimination from the body
-
Transamination
-
Removal of nitrogen from amino acids
-
Urea Cycle
-
Glucose-alanine cycle (Cahill Cycle)
-
NAG
-
Glutamate dehydrogenase
-
Alanine aminotransferase (ALT)
-
Aspartate aminotransferase (AST)
-
Glutamate synthetase
-
Glutaminase
-
Carbamoyl phosphate synthase I
-
Ornithine transcarbamylase
-
Cystinuria
Describe how inherited and acquired deficiencies in the urea cycle can cause hyperammonemia
-
Ornithine transcarbamylase disorder
-
Carbamoyl phosphate synthetase I deficiency
-
N-acetylglutamate synthase deficiency (not congenital, but something to know)
Describe the concept of the amino acid pool and compare and contrast amino acids acquisition from endogenous and exogenous protein sources
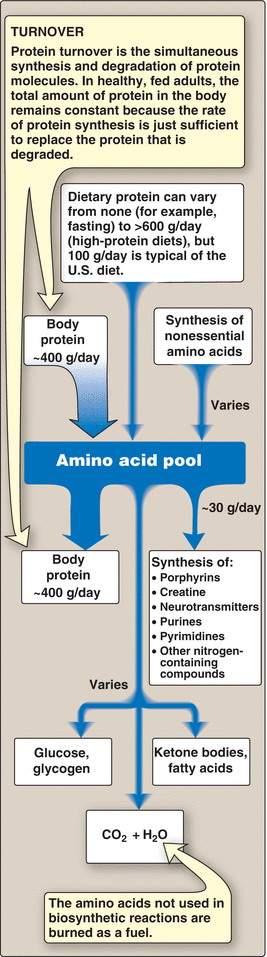
Think about the endogenous (i.e., body proteins) versus exogenous (dietary protein) and how amino acids are liberated from these proteins (proteasome degradation versus the use of enzymes during dietary digestion.)
Describe ammonia transfer to and from amino acids and its transport and subsequent elimination from the body
The Urea Cycle is a very high-yield thing to remember. Therefore, Osmosis has a video on it.
Describe how inherited and acquired deficiencies in the urea cycle can cause hyperammonemia
|
Ornithine transcarbamylase deficiency |
Genetic |
Most common urea-cycle defect, leading to elevated blood ammonia and orotic acid levels and will lead to mental impairment if not treated
|
|
CPSI deficiency, argininosuccinate synthetase deficiency, argininosuccinate lyase deficiency, and arginase deficiency |
All genetic |
Mutations in urea-cycle enzymes, leading to various degrees of hyperammonemia and inability to synthesize urea; can be distinguished by the type of urea-cycle intermediates that accumulate in the blood
|