


- Suggested reading
- Reading guide
- Need to Know
There are two chapters from Lippincott for suggested readings. These two chapters are pretty succinct for the topics at hand. Chapter 10 is on gluconeogenesis and Chapter 11 is on glycogen metabolism. The short descriptions listed below are some of the very big-picture, high-yield points. The reading and the class session will go into more depth and detail. The reading guides are also posted. As I said in other modules, this reading guide is for people who have a difficult time focusing on these types of chapters. Most students DO NOT need to use the guide.
Diagram and describe gluconeogenesis, including the rate-determining enzymes and the conversion of glycerol, lactate, and certain amino acids into glucose
- Gluconeogenesis
- Pyruvate carboxylase
- PEPCK
- Fructose-1,6-bisphosphatase
Diagram and describe how the Cori cycle operates and its significance
- Cori Cycle
Diagram and describe how glycogen is synthesized and degraded, including the rate-determining enzymes
- Glycogen synthesis and glycogenolysis
- Glycogen structure
- Glycogenin
- Glycogen synthase
- Branching enzyme
- Glucose-6-phosphatase
- Glycogen Phosphorylase
- Phosphoglucomutase
Identify the enzyme deficiency for glycogen storage diseases, types I, II, III, and V, and contrast the clinical features and prognoses
-
Glycogen storage diseases (I, II, III, IV, V, and VI)
Diagram and describe gluconeogenesis, including the rate-determining enzymes and the conversion of glycerol, lactate, and certain amino acids into glucose
Watch this Osmosis video on gluconeogenesis.
Diagram and describe how the Cori cycle operates and its significance
This is covered quite well in the text, and I’ll go over it in class. The picture below sums it up quite well.
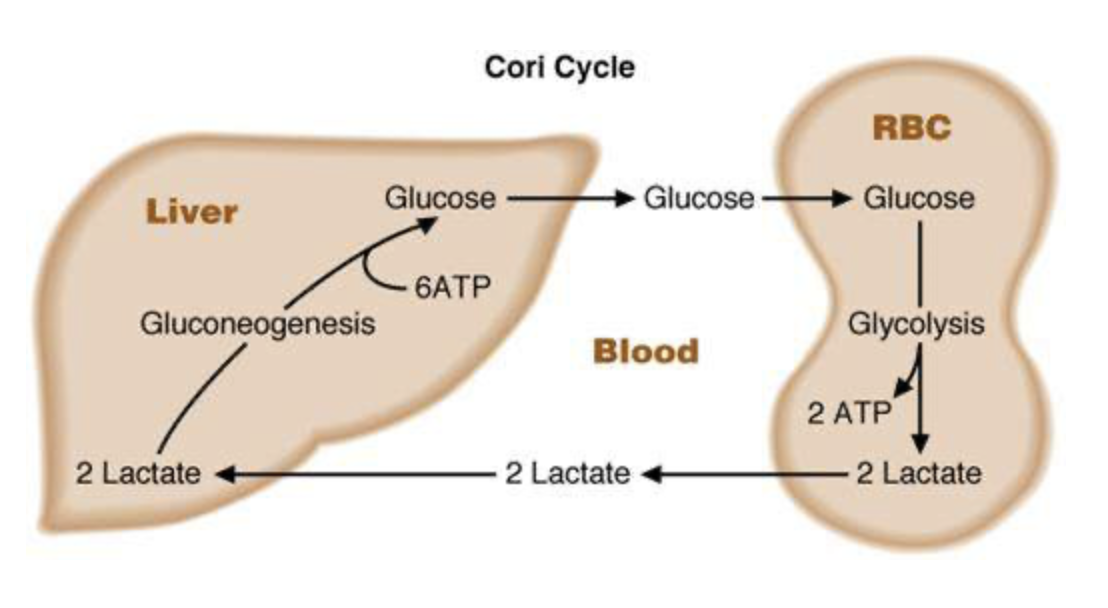
If you are looking for a video to help you remember it, try this Pixorize video (hey, it’s something a bit different!).
Diagram and describe how glycogen is synthesized and degraded, including the rate-determining enzymes
Watch this Osmosis video on glycogen.
Identify the enzyme deficiency for glycogen storage diseases, types I, II, III, and V, and contrast the clinical features and prognoses
| Type | Enzyme affected | Primary organ involved | Manifestations |
|---|---|---|---|
|
O |
Glycogen synthase
|
Liver |
Hypoglycemia, hyperketonemia, failure to thrive, early death
|
|
Ib |
Glucose 6-phosphatase ( von Gierke disease) |
Liver |
Enlarged liver and kidney, growth failure, severe fasting hypoglycemia, acidosis, lipemia, thrombocyte dysfunction
|
|
II |
Lysosomal α-glucosidase ( Pompe disease): may see clinical symptoms in childhood, juvenile, or adult life stages, depending on the nature of the mutation
|
All organs with lysosomes |
Infantile form: Early-onset progressive muscle hypotonia, cardiac failure, death before age 2 years. Juvenile form: Later-onset myopathy with variable cardiac involvement. Adult form: limb-girdle muscular dystrophy–like features. Glycogen deposits accumulate in lysosomes. |
|
III |
Amylo-1,6-glucosidase (debrancher): form IIIa is the liver and muscle enzymes, form IIIb is a liver-specific form, and IIIc a muscle-specific form
|
Liver, skeletal muscle, heart
|
Fasting hypoglycemia; hepatomegaly in infancy and some myopathic features. Glycogen deposits have short outer branches.
|
|
IV |
Amylo-4,6-glucosidase (branching enzyme) (Andersen disease)
|
Liver |
Hepatosplenomegaly; symptoms may arise from a hepatic reaction to the presence of a foreign body (glycogen with long outer branches); usually fatal
|
|
V |
Muscle glycogen phosphorylase (McArdle disease) (expressed as either adult or infantile form)
|
Skeletal muscle
|
Exercise-induced muscular pain, cramps, and progressive weakness, sometimes with myoglobinuria
|
|
VIc |
Liver glycogen phosphorylase (Hers disease) and its activating system (includes mutations in liver phosphorylase kinase and liver protein kinase A)
|
Liver |
Hepatomegaly, mild hypoglycemia; good prognosis
|
|
VII |
Phosphofructokinase-1 (Tarui syndrome)
|
Muscle, red blood cells
|
As in type V; in addition, enzymopathic hemolysis
|
|
XI |
GLUT 2 (glucose/galactose transporter); Fanconi-Bickel syndrome
|
Intestine, pancreas, kidney, liver
|
Glycogen accumulation in liver and kidney; rickets, growth retardation, glucosuria |
aAll of these diseases except type O are characterized by increased glycogen deposits.
bGlucose 6-phosphatase is composed of several subunits that also transport glucose, glucose 6-phosphate, phosphate, and pyrophosphate across the endoplasmic reticulum membranes. Therefore, there are several subtypes of this disease, corresponding to defects in the different subunits. Type Ia is a lack of glucose 6-phosphatase activity, type Ib is a lack of glucose 6-phosphate translocase activity, type Ic is a lack of phosphotranslocase activity, and type Id is a lack of glucose translocase activity.
cGlycogen storage diseases IX (hepatic phosphorylase kinase) and X (hepatic protein kinase A) have been reclassified to VI, which now refers to the hepatic glycogen phosphorylase activating system.
Sources: Parker PH, Ballew M, Greene HL. Nutritional management of glycogen storage disease. Annu Rev Nutr. 1993;13:83–109. Copyright © 1993 by Annual Reviews, Inc.; Shin YS. Glycogen storage disease: clinical, biochemical, and molecular heterogeneity. Semin Pediatr Neurol. 2006;13:115–120; Ozen H. Glycogen storage diseases: new perspectives. World J Gastroenterol. 2007;13:2541–2553.